1. シミュレーションの準備¶
タンパク質(Lysozyme)のシミュレーションを行うにあたって、まずは対象とする系を作成する必要がある。 系の作成は次の3つのステップで行う。
タンパク質の構造を準備
シミュレーションボックスの作成と溶媒の追加
イオンの追加
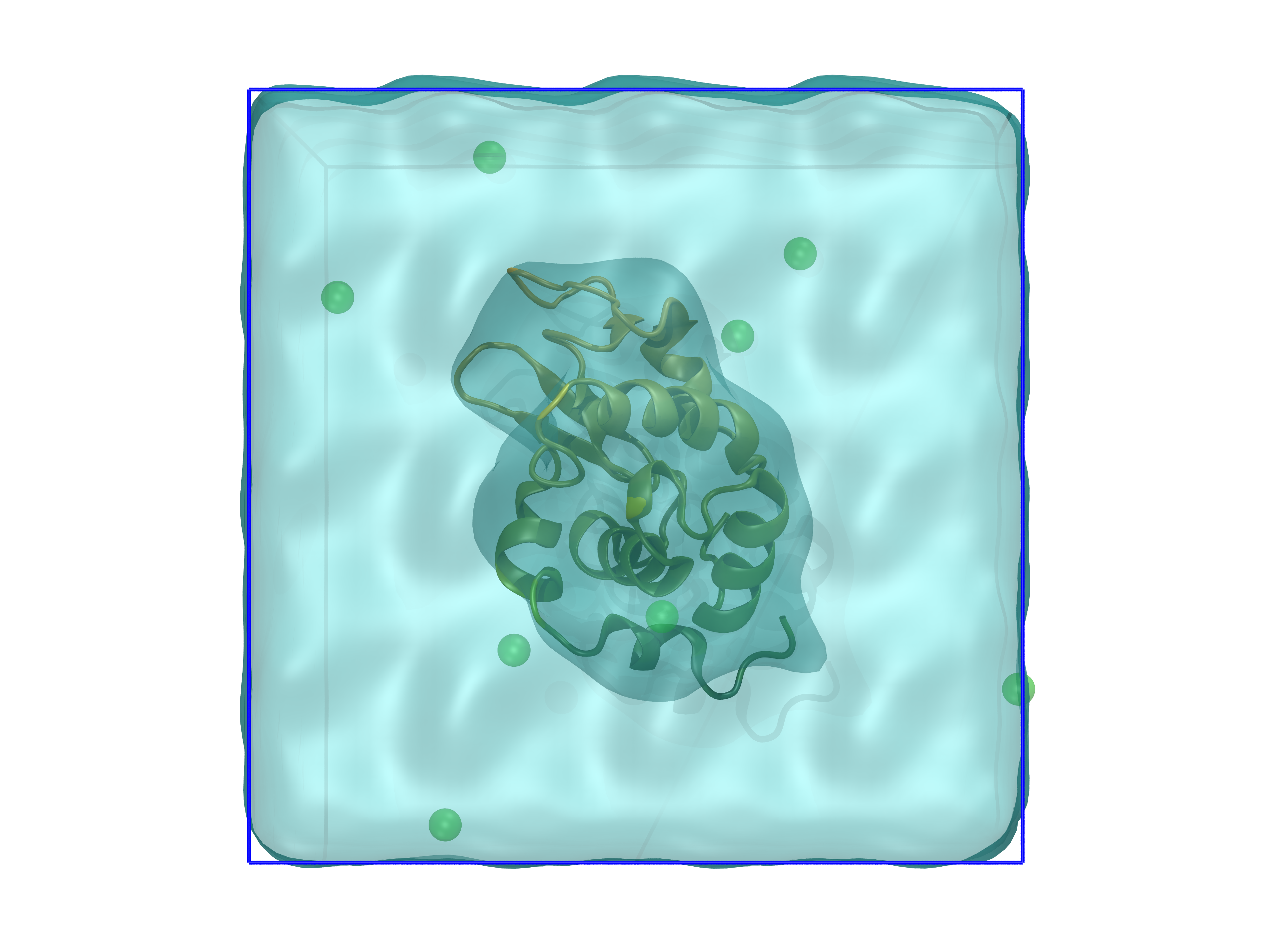
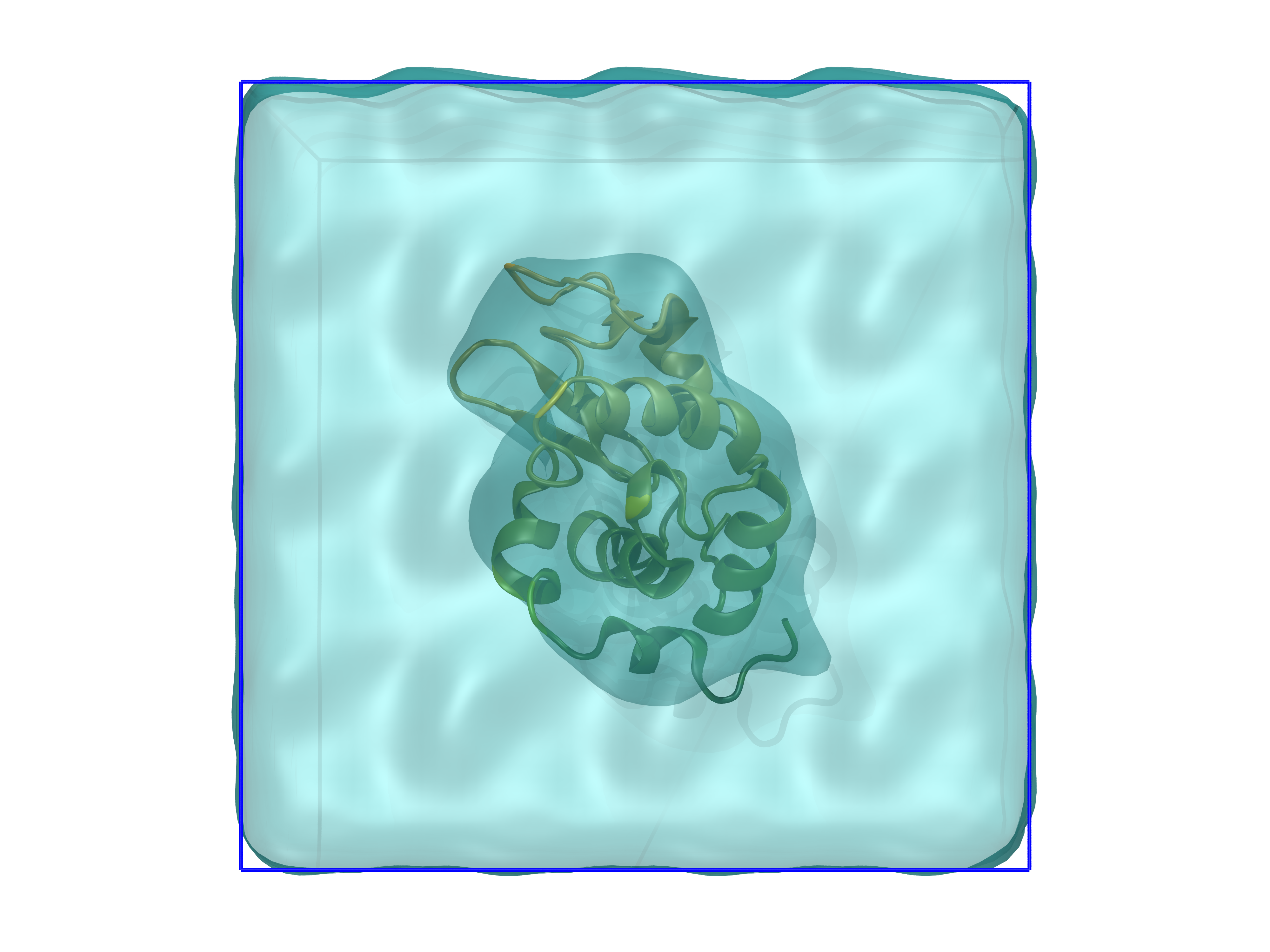
今回作成する系は図のようになる。 水とイオンが入ったボックスにタンパク質が入るような構造を作成する。
タンパク質の構造を準備¶
まず、タンパク質の構造ファイルをダウンロードする必要がある。 RCSBのサイトからlysozyme(PDBコード:1AKI)を検索し、PDBファイルをダウンロードする。 サイトからダウンロードできない場合はここからダウンロードしてほしい。 このファイルをVMDで確認すると、タンパク質とその周りの結晶水の構造を確認することができる。
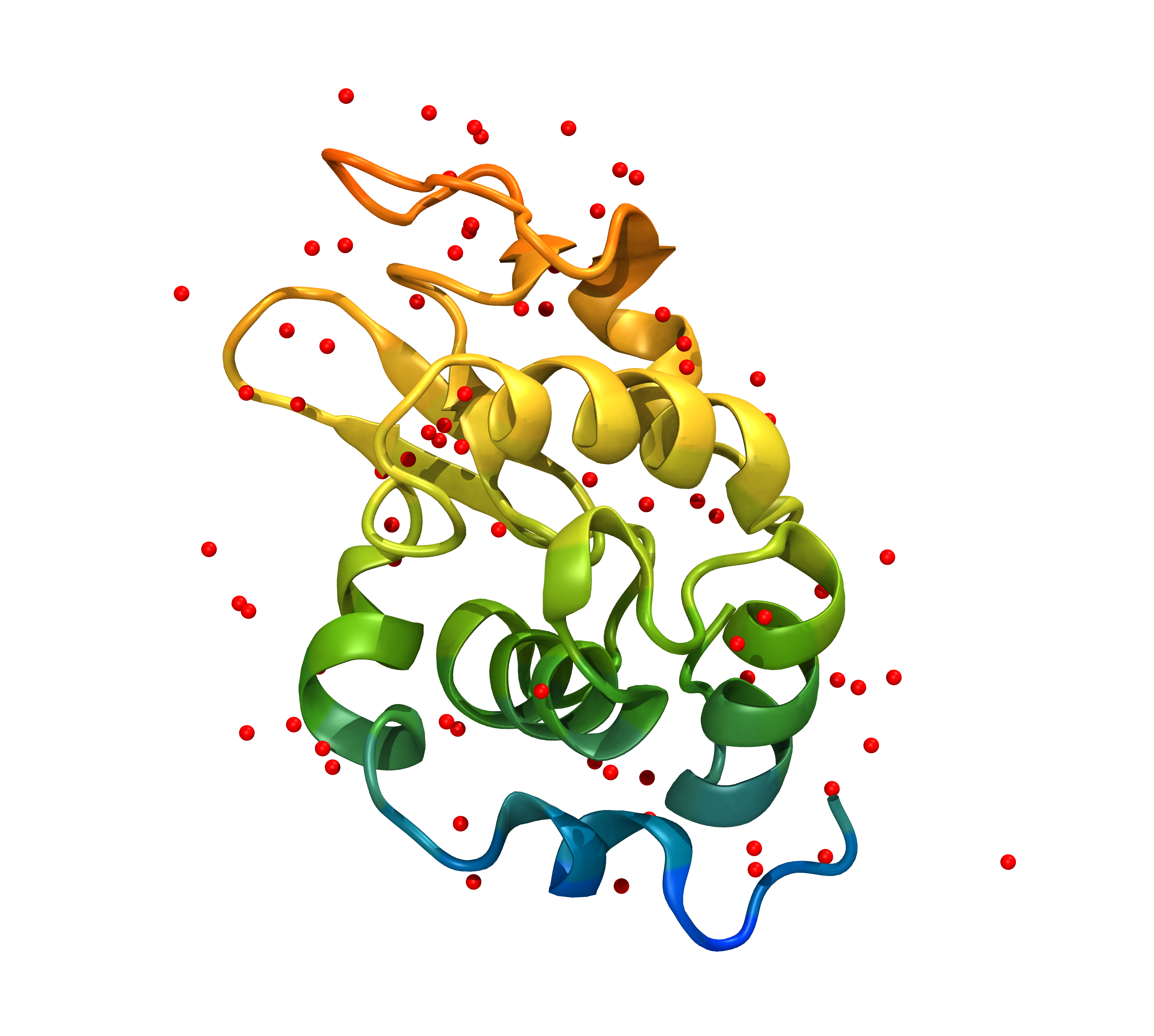
今回は結晶水の構造は必要ないので、PDBファイルのHOHを含む行をgrep -vで除いておく。
grep -v HOH 1aki.pdb > 1AKI_clean.pdb
これで新しく作成したファイルをVMDで確認すると、結晶水が取り除かれている。
Before : 1aki.pdb
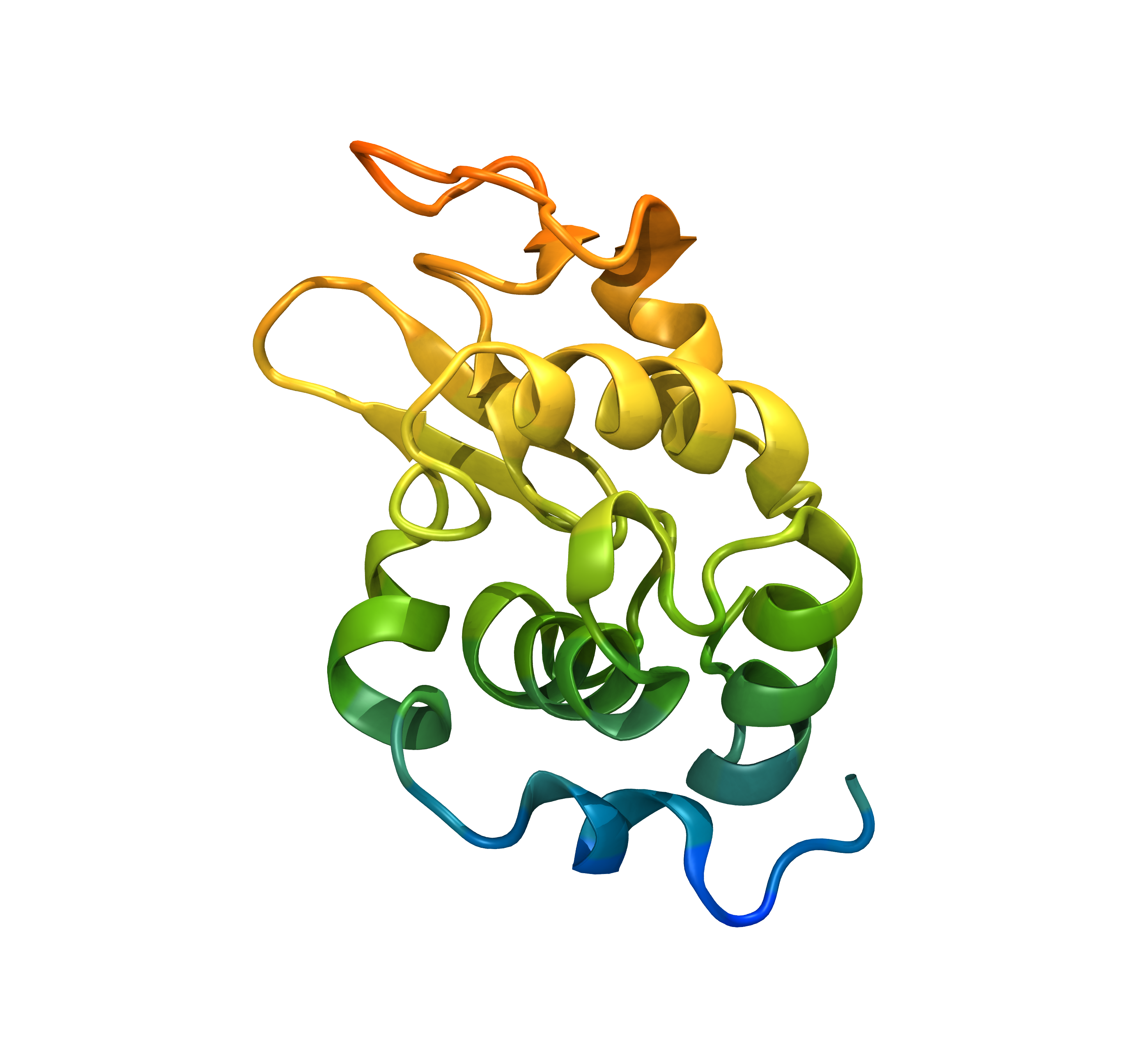
After : 1AKI_clean.pdb
次に、タンパク質の構造ファイルからMD計算に使用する以下のファイルを作成する。
トポロジーファイル(topol.top)
位置拘束ファイル(posre.itp)
GROMACSフォーマットの構造ファイル(1AKI_processed.gro)
これらのファイルはgmx pdb2gmxを使用してPDBファイルから一括で作成できる。 -waterを指定することで、計算で使用する水モデルの種類を指定することができる。
| gmx pdb2gmxオプション | 説明 |
|---|---|
| -f | 入力pdbファイルの指定 |
| -o | 出力groファイルの指定 |
| -water | 水モデルの指定(今回はSPE/C) |
gmx pdb2gmx -f 1AKI_clean.pdb -o 1AKI_processed.gro -water spce
コマンドを実行すると使用する力場を聞かれるので、今回はOPLS-AAを選択する。15と入力してEnterキーを押す。
Select the Force Field:
From '/usr/local/gromacs/share/gromacs/top':
1: AMBER03 protein, nucleic AMBER94 (Duan et al., J. Comp. Chem. 24, 1999-2012, 2003)
2: AMBER94 force field (Cornell et al., JACS 117, 5179-5197, 1995)
3: AMBER96 protein, nucleic AMBER94 (Kollman et al., Acc. Chem. Res. 29, 461-469, 1996)
4: AMBER99 protein, nucleic AMBER94 (Wang et al., J. Comp. Chem. 21, 1049-1074, 2000)
5: AMBER99SB protein, nucleic AMBER94 (Hornak et al., Proteins 65, 712-725, 2006)
6: AMBER99SB-ILDN protein, nucleic AMBER94 (Lindorff-Larsen et al., Proteins 78, 1950-58, 2010)
7: AMBERGS force field (Garcia & Sanbonmatsu, PNAS 99, 2782-2787, 2002)
8: CHARMM27 all-atom force field (CHARM22 plus CMAP for proteins)
9: GROMOS96 43a1 force field
10: GROMOS96 43a2 force field (improved alkane dihedrals)
11: GROMOS96 45a3 force field (Schuler JCC 2001 22 1205)
12: GROMOS96 53a5 force field (JCC 2004 vol 25 pag 1656)
13: GROMOS96 53a6 force field (JCC 2004 vol 25 pag 1656)
14: GROMOS96 54a7 force field (Eur. Biophys. J. (2011), 40,, 843-856, DOI: 10.1007/s00249-011-0700-9)
15: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)
これで必要なファイルが作成されているはずだ。 なお、作成したトポロジーファイルの詳細については4.【補足】トポロジーファイルの詳細を参照してほしい。
シミュレーションボックスの作成と溶媒の追加¶
gmx pdb2gmxで作成したGROMACSフォーマットの構造ファイル(1AKI_processed.gro)にはまだタンパク質の構造しか含まれていない。 そこで次にタンパク質が入るようなシミュレーションボックスを作成し、その中を溶媒で満たす操作を行う。
シミュレーションボックスの作成はgmx editconfで行う。今回、シミュレーションボックスには周期的境界条件を満たす立方体を用いる。 この時、タンパク質とボックスの境界との距離を1.0 nm以上空けることで、周期的境界条件では隣のタンパク質との距離を2.0 nm以上確保することができる。 いくつかのオプションを指定してコマンドを実行する。
| gmx editconfオプション | 説明 |
|---|---|
| -f | 入力構造ファイルの指定 |
| -o | 出力構造ファイルの指定 |
| -c | タンパク質をボックスの中央に配置 |
| -d | タンパク質とボックス境界との距離を指定 |
| -bt | ボックスタイプの指定 |
gmx editconf -f 1AKI_processed.gro -o 1AKI_newbox.gro -c -d 1.0 -bt cubic
生成した1AKI_newbox.groをVMDで確認すると、シミュレーションボックスが定義されている。
ボックスが定義できたので、続いてgmx solvateにより溶媒で系を満たす。 今回の溶媒は水であり、SPCモデルを用いている。 SPCの構造ファイル(spc216.gro)はGROMACSに標準でインストールされているので、特別にファイルを用意する必要はない。
| gmx solvateオプション | 説明 |
|---|---|
| -cp | タンパク質(溶質)構造ファイルの指定 |
| -cs | 溶媒(SPC水モデル)構造ファイルの指定 |
| -o | 出力構造ファイルの指定 |
| -p | トポロジーファイルの指定 |
gmx solvate -cp 1AKI_newbox.gro -cs spc216.gro -o 1AKI_solv.gro -p topol.top
これでボックス内を溶媒で満たすことができた。VMDで確認すると、水分子がタンパク質の周りに配置されている様子が見える。
また、gmx solvateではオプションにトポロジーファイル(topol.top)を指定している。 溶媒の追加によりトポロジーファイルの[ molecules ]のセクションが更新されており、SOL(溶媒)が記述されていることがわかるだろう。
topol.top
[ molecules ]
; Compound #mols
Protein_A 1
SOL 10832
イオンの追加¶
さらに、系に対してイオンを追加する。 トポロジーファイルを確認するとわかるが、タンパク質(lysozyme)の電荷は+8(qtot 8)であるため、イオンを加えて電荷を打ち消す必要がある。 イオンの追加にはgmx genionを用いるが、その入力ファイルには系の全原子のパラメータを含む形式であるtprファイルを作成しなければならない。 tprファイルは、系の構造ファイルとトポロジーファイルの両方を使用することでgmx gromppによって作成することができる。
加えて、gmx gromppを実行するためにはさらに追加でmdpファイル(molecular dynamics parameter file)を用意する必要がある。 mdpファイルはMD計算で使用するパラメータの設定ファイルである。 mdpファイルは通常、MD計算の条件(温度、圧力、タイムステップなど)を記述し、gmx gromppによって実際にMD計算を行う際の入力ファイル(tprファイル)を作成するために使用される。 ただし現時点ではイオンを追加するだけなので、今回使用するmdpファイルはその設定のみが記述されている。
ions.mdpをここからダウンロードしてgmx gromppを実行する。
| gmx gromppオプション | 説明 |
|---|---|
| -f | 入力mdpファイルの指定 |
| -c | 入力構造ファイルの指定 |
| -p | トポロジーファイルの指定 |
| -o | 出力tprファイルの指定 |
gmx grompp -f ions.mdp -c 1AKI_solv.gro -p topol.top -o ions.tpr
tprファイルは系の全原子の情報を記述したバイナリファイルであり、これを用いて系にイオンを追加する。
| gmx genionオプション | 説明 |
|---|---|
| -s | 入力tprファイルの指定 |
| -o | 出力構造ファイルの指定 |
| -p | トポロジーファイルの指定 |
| -pname | 追加する陽イオンの指定 |
| -nname | 追加する陰イオンの指定 |
| -neutral | 系全体の電荷が0になるようにイオンを追加 |
gmx genion -s ions.tpr -o 1AKI_solv_ions.gro -p topol.top -pname NA -nname CL -neutral
実行すると溶媒として利用する分子を聞かれるので、13(SOL)を選択する。 これにより、溶媒の水分子の一部をイオンに置き換えることができる。 今回は+8の電荷を打ち消すために8つのClイオンが追加されており、トポロジーファイルを確認すると[ molecules ]にイオンが反映されている。
topol.top
[ molecules ]
; Compound #mols
Protein_A 1
SOL 10636
CL 8
以上で、系の構築は完了である。VMDで確認してみると、下図のような構造が見られるであろう。
まとめ¶
これまでの内容をまとめる。系の構築のために以下の手順を踏んだ。
タンパク質のPDBファイルを
gmx pdb2gmxによりGROMACSフォーマットに変換した。シミュレーションボックスを
gmx editconfで作成し、gmx solvateによりボックス内を溶媒で満たした。ボックス内に
gmx genionでイオンを追加した。
次回は作成した系を用いて実際にMD計算をしていく。