3. mdpファイルの作成とシミュレーションの実行¶
前回まででアルコール二重膜系を作成し、CHARMM36力場を使用したGROMACSでのシミュレーションを開始できるまでに至った。 今回は実際にMDシミュレーションを行うが、シミュレーションの設定をメインに解説し、好きな条件でシミュレーションを実行できるようになることを目標とする。 シミュレーションの手順はチュートリアル1と同様、下記のように進めていく。
エネルギー最小化(00_em)
NVT平衡化(01_nvt)
NPT平衡化(02_npt)
本計算(03_npt)
また、このページで解説したmdpファイルの設定について、より詳しい解説は【GROMACS.mdpオプション】のページで紹介している。 GROMACSの操作に慣れてきたら参考にしてほしい。
エネルギー最小化(00_em)¶
まずは、作成した構造のエネルギー最小化を行う。 設定条件を記述したmdpファイルは次のようになる。
00_em.mdp
integrator = steep
emtol = 1000.0
emstep = 0.01
nsteps = 50000
nstlist = 10
cutoff-scheme = Verlet
ns_type = grid
coulombtype = PME
rcoulomb = 1.2
rvdw = 1.2
pbc = xyz
エネルギー最小化には最急降下法を用いるのでintegrator = steepとし、系の最大力Fmaxが1000 kJ mol-1 nm-1以下となるまで緩和を行う。 この時、同時にステップ間隔と最大ステップ数を指定している。
また、次に非結合相互作用(LJ相互作用、クーロン相互作用)に関する設定している。 近接原子の定義、探し方、更新頻度などを設定した後、それらが遠距離の場合と近距離の場合で異なる相互作用の取り扱いをしている。 coulombtype = PMEは遠距離クーロン相互作用にパーティクルメッシュエワルド法を使用する設定である。 また、rcoulombとrvdwは近距離クーロン相互作用及び近距離LJ相互作用のカットオフ距離である。
最後、pbc = xyzは周期的境界条件を3次元に適用している。
このmdpファイルと作成した二重膜系の構造を用いてgmx gromppとgmx mdrunを実行する。
gmx grompp -f 00_em.mdp -c system.gro -p topol.top -o 00_em.tpr
gmx mdrun -deffnm 00_em
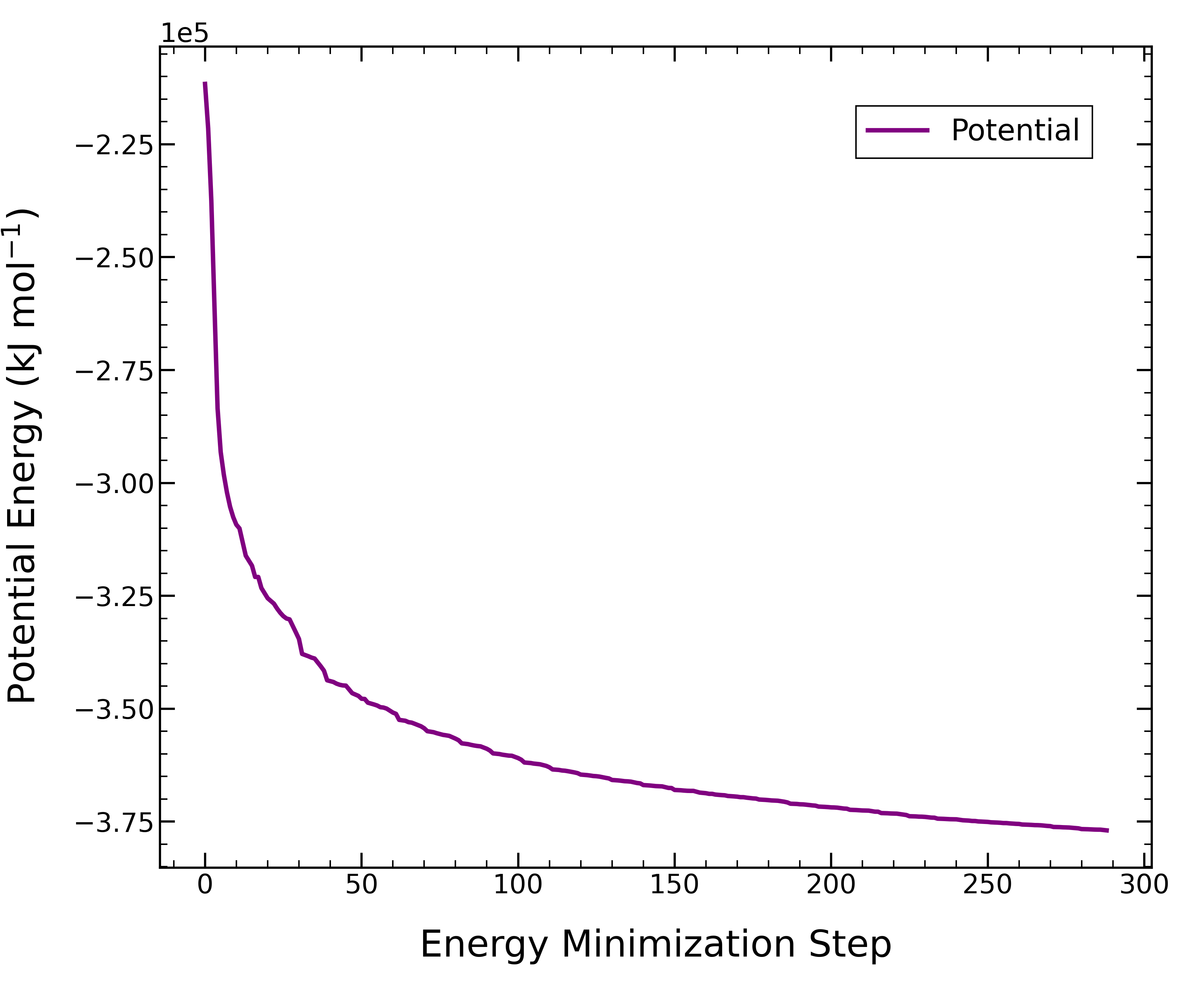
計算が終了すると、エネルギー最小化された系の構造が得られる。 エネルギー最小化過程のポテンシャルエネルギー変化を出力してみた。
gmx energy -f 00_em.tpr -o potential.xvg
NVT平衡化(01_nvt)¶
エネルギー最小化ができたら、NVT平衡化を行う。 mdpファイルには温度制御の設定を記述する必要があり、またシミュレーションの長さに合わせてステップ数を決定する。 今回はNose-Hooverの方法で温度制御を行い、310.15 Kで125.0 psのNVT計算を行う設定をした。
01_nvt.mdp
integrator = md
dt = 0.001
nsteps = 125000
nstxtcout = 5000
nstxout = 5000
nstvout = 5000
nstfout = 5000
nstcalcenergy = 100
nstenergy = 1000
nstlog = 1000
cutoff-scheme = Verlet
nstlist = 20
rlist = 1.2
coulombtype = PME
rcoulomb = 1.2
vdwtype = Cut-off
vdw-modifier = Force-switch
rvdw_switch = 1.0
rvdw = 1.2
tcoupl = Nose-Hoover
tc_grps = SYSTEM
tau_t = 1.0
ref_t = 310.15
constraints = h-bonds
constraint_algorithm = LINCS
nstcomm = 100
comm_mode = linear
comm_grps = SYSTEM
gen-vel = yes
gen-temp = 310.15
gen-seed = -1
refcoord_scaling = com
上から順番に見ていく。まず、MD計算を行うのでintegrator = mdとし、タイムステップはdt = 0.001としている。 このタイムステップの単位はpsなので、設定したタイムステップは1 fsとなる。 nsteps = 125000とすれば、0.001 ps × 125000 = 125.0 psのシミュレーションが実行できる。
次に出力情報の設定をしている。 主にトラジェクトリファイルを何ステップごとに記述するかを設定しており、nstxout、nstvout、nstfoutはそれぞれ系の原子の座標、速度及び働く力に対応する。 これらの情報はtrrファイルにトラジェクトリとして出力されるが、nstxtcoutを指定するとxtcファイルにも座標情報を出力することができる。 また、エネルギーや計算ログの出力もnstenergyとnstlogで制御できる。
温度制御はtcoupl = Nose-Hooverを適用している。 ref_tが参照温度なので、ここにシミュレーションしたい温度である310.15 Kを指定する。
constraintsでは結合の拘束条件とそのアルゴリズムを指定する。 今回は水素結合に対してLINCSという方法で結合拘束をしている。
comm_modeは系の質量中心が並進運動してしまう場合に、その速度を取り除く設定である。 nstcommでその操作の頻度を指定する。
最後、gen-velで系の原子に対して初速度を与える。 初速度はマクスウェル分布に従って生成され、温度はgen-tempで与えることができる。 普通は温度制御時の参照温度と同じ値になるであろう。
設定が確認できたらgmx gromppとgmx mdrunを実行する。
gmx grompp -f 01_nvt.mdp -c 00_em.gro -p topol.top -o 01_nvt.tpr
gmx mdrun -deffnm 01_nvt
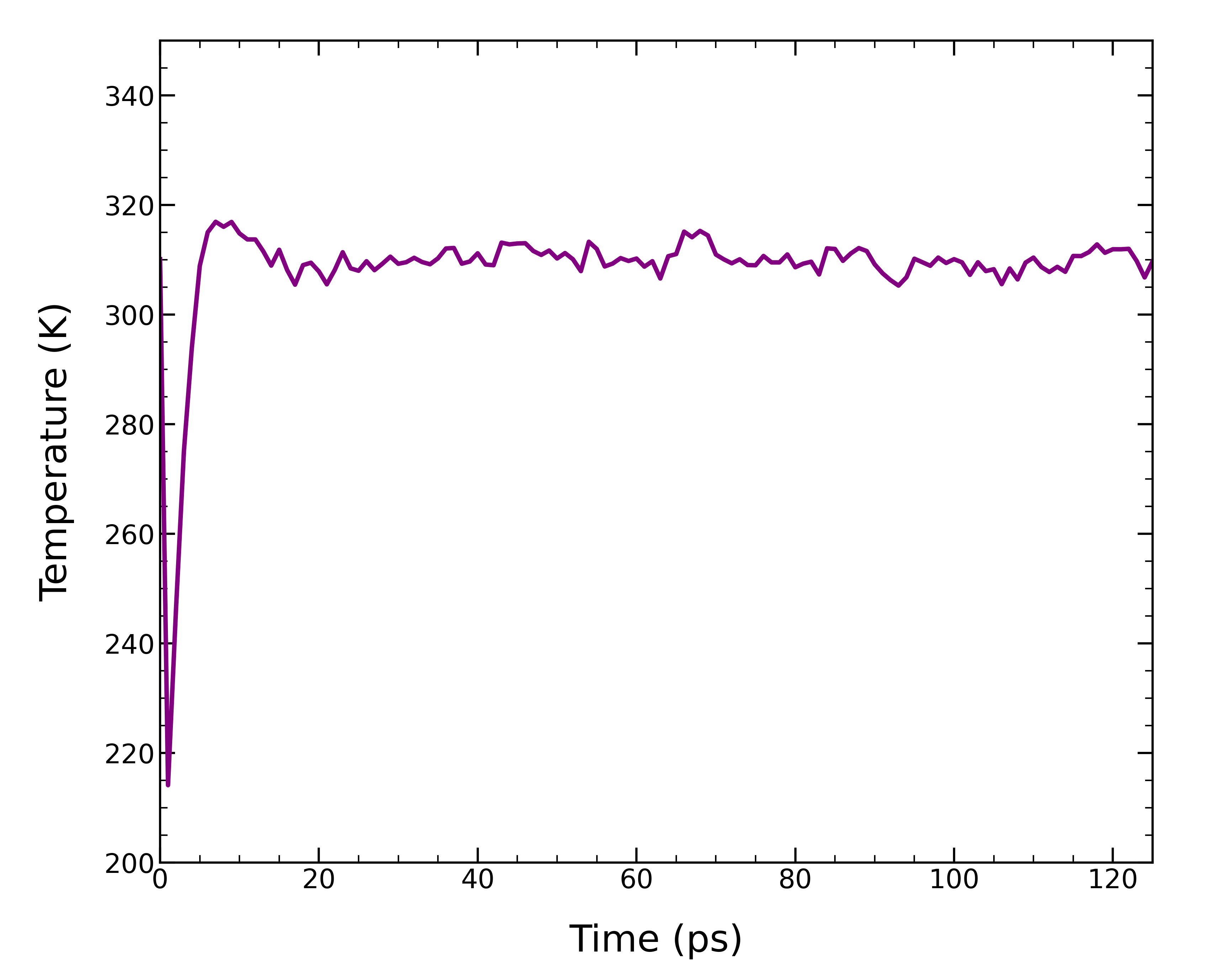
チュートリアル①と同様に、NVT平衡化で温度が一定となっているかを確認する。 グラフにプロットすると、125.0 psの間温度が安定していることがわかる。
gmx energy -f 01_nvt.tpr -o temperature.xvg
NPT平衡化(02_npt)¶
系の温度が安定したら、続いてNPT平衡化を行う。 ここでの設定はほとんど本計算と同じものになるが、圧力が安定するまでがこのステップの役割である。 圧力制御にはParrinello-Rahmanの方法を使用し、参照圧力1.0 barとして500.0 psのシミュレーションを行う。
02_npt.mdp
integrator = md
dt = 0.002
nsteps = 250000
nstxtcout = 5000
nstvout = 5000
nstfout = 5000
nstcalcenergy = 100
nstenergy = 1000
nstlog = 1000
cutoff-scheme = Verlet
nstlist = 20
rlist = 1.2
coulombtype = pme
rcoulomb = 1.2
vdwtype = Cut-off
vdw-modifier = Force-switch
rvdw_switch = 1.0
rvdw = 1.2
tcoupl = Nose-Hoover
tc_grps = SYSTEM
tau_t = 1.0
ref_t = 310.15
pcoupl = Parrinello-Rahman
pcoupltype = semiisotropic
tau_p = 5.0
compressibility = 4.5e-5 4.5e-5
ref_p = 1.0 1.0
constraints = h-bonds
constraint_algorithm = LINCS
continuation = yes
nstcomm = 100
comm_mode = linear
comm_grps = SYSTEM
refcoord_scaling = com
おおよその設定はNVT平衡化のものと同様であるが、圧力制御のセクションが追加されている。 圧力制御はpcoupl = Parrinello-Rahmanとし、参照圧力はref_pに1.0 barを指定する。 ここで注意するのは、圧力の制御タイプがpcoupltype = semiisotropicとなっている点である。 今回のシミュレーションはアルコールの二重膜を扱っており、膜はxy方向に広がっているので、xy方向とz方向で同じ圧力制御をすることができない。 従って、制御タイプにはsemiisotropic(半等方性)を設定し、ref_pにはxy方向とz方向でそれぞれの参照圧力を与える。
また、NPT平衡化はNVT平衡化から継続するのでcontinuation = yesとし、ここからタイムステップはdt = 0.002としている。
同様に、gmx gromppとgmx mdrunを実行する。
gmx grompp -f 02_npt.mdp -c 01_nvt.gro -p topol.top -o 02_npt.tpr
gmx mdrun -deffnm 02_npt
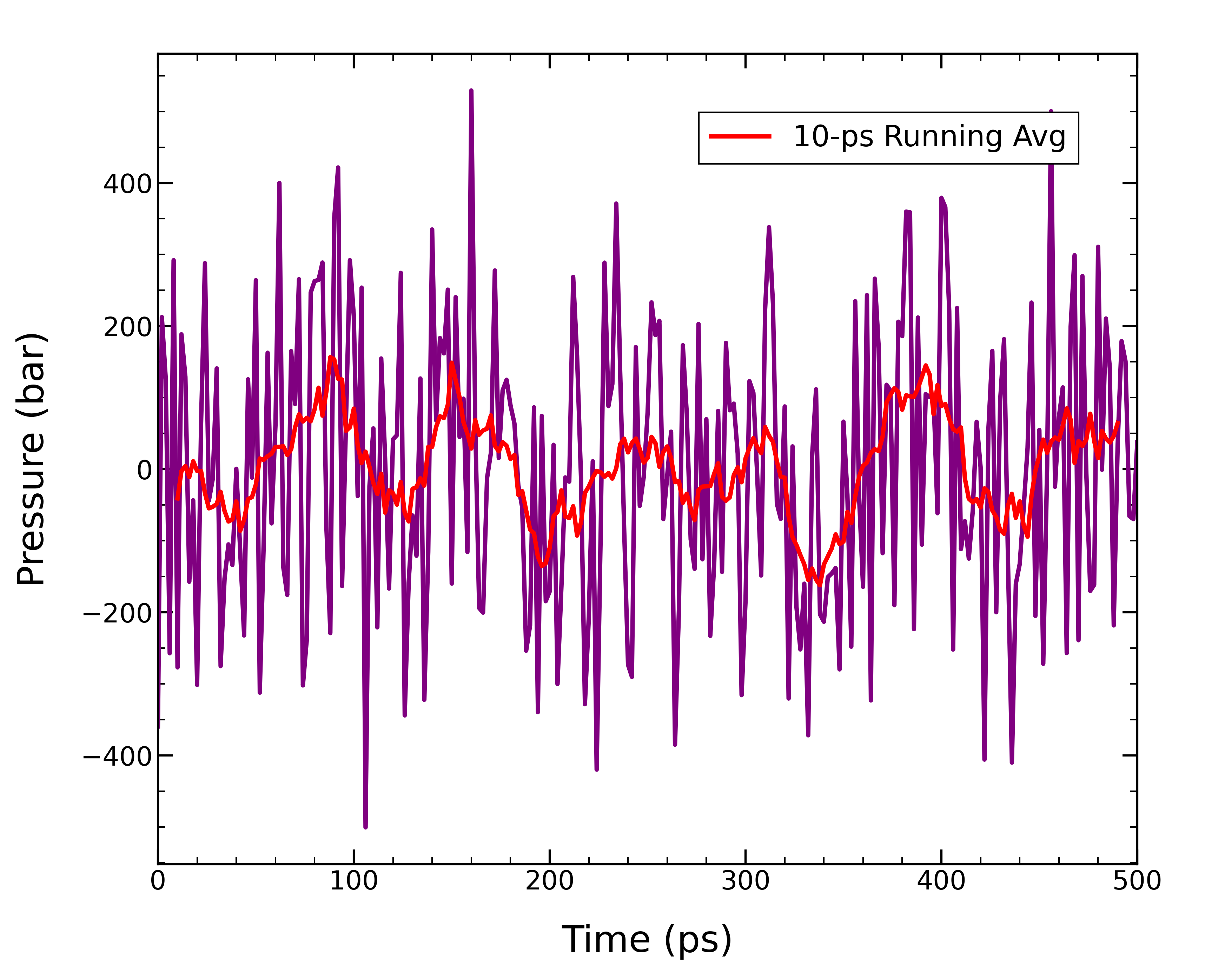
シミュレーションが完了すると、310.15 K、1.0 barでのアルコール二重膜の構造が得られる。 トラジェクトリを確認すると、膜分子の占有面積が小さくなり、系の大きさが変化したことが見られる。 圧力についてもグラフにプロットしてみた。
gmx energy -f 02_npt.tpr -o pressure.xvg
本計算(03_npt)¶
最後は本計算を行う。 本計算はNPT平衡化の延長なので、ステップ数を増やして50.0 nsのシミュレーションを行う。 ここまでで温度と圧力が十分に安定していることを確認して、シミュレーションを実行しよう。
03_npt.mdp
integrator = md
dt = 0.002
nsteps = 25000000
nstxtcout = 50000
nstvout = 50000
nstfout = 50000
nstcalcenergy = 100
nstenergy = 1000
nstlog = 1000
cutoff-scheme = Verlet
nstlist = 20
rlist = 1.2
coulombtype = pme
rcoulomb = 1.2
vdwtype = Cut-off
vdw-modifier = Force-switch
rvdw_switch = 1.0
rvdw = 1.2
tcoupl = Nose-Hoover
tc_grps = SYSTEM
tau_t = 1.0
ref_t = 310.15
pcoupl = Parrinello-Rahman
pcoupltype = semiisotropic
tau_p = 5.0
compressibility = 4.5e-5 4.5e-5
ref_p = 1.0 1.0
constraints = h-bonds
constraint_algorithm = LINCS
continuation = yes
nstcomm = 100
comm_mode = linear
comm_grps = SYSTEM
refcoord_scaling = com
実行コマンドも変わらず、gmx gromppとgmx mdrunで実行する。
gmx grompp -f 03_npt.mdp -c 02_npt.gro -p topol.top -o 03_npt.tpr
gmx mdrun -deffnm 03_npt
シミュレーションには時間がかかると思うが、計算が終了すると次のような構造が見られるであろう。 二重膜を形成するアルコール分子が一定方向に傾き、ラメラ構造を形成している。

まとめ¶
チュートリアル②は以上となる。学習内容をまとめると以下のようになる。
CHARMM-GUIなどを使用して、系の構造の作成方法を学習した。GROMACSへの力場の導入方法、及び分子の取り扱い方を理解した。
mdpファイルを編集し、目的のシミュレーション条件に合った設定をした。
ここまでできればGROMACSは一通り扱えるようになったのではないだろうか。 これから独自のシミュレーションを行い、さらに深い知識を身に付けていってほしい。 また、GROMACSのチュートリアルページは備忘録としても活用してほしい。